
Abstract
Since 1987, keratinocytes have been cultured at the Queen Astrid Military Hospital. These keratinocytes have been used routinely as auto and allografts on more than 1,000 patients, primarily to accelerate the healing of burns and chronic wounds. Initially the method of Rheinwald and Green was used to prepare cultured epithelial autografts, starting from skin samples from burn patients and using animal-derived feeder layers and media containing animal-derived products. More recently we systematically optimised our production system to accommodate scientific advances and legal changes. An important step was the removal of the mouse fibroblast feeder layer from the cell culture system. Thereafter we introduced neonatal foreskin keratinocytes (NFK) as source of cultured epithelial allografts, which significantly increased the consistency and the reliability of our cell production. NFK master and working cell banks were established, which were extensively screened and characterised. An ISO 9001 certified Quality Management System (QMS) governs all aspects of testing, validation and traceability. Finally, as far as possible, animal components were systematically removed from the cell culture environment. Today, quality controlled allograft production batches are routine and, due to efficient cryopreservation, stocks are created for off-the-shelf use. These optimisations have significantly increased the performance, usability, quality and safety of our allografts. This paper describes, in detail, our current cryopreserved allograft production process.
Similar content being viewed by others

Introduction
In the late 1970s, Rheinwald and Green described a method for serial cultivation of human epidermal keratinocytes on a feeder layer of mouse fibroblasts in a mitogen-rich medium containing fetal bovine serum (Rheinwald and Green 1975, 1977). O’Connor et al. (1981) reported the use of autologous keratinocyte cultures for the treatment of burns, and over time an increasing number of culture and delivery systems of epithelial grafts were developed, mainly in the burn care field (Madden et al. 1986; Herzog et al. 1988; Burt et al. 1989; Compton et al. 1990; De Luca et al. 1989; Bolivar-Flores et al. 1990; Fratianne et al. 1993; Phillips et al. 1993; Rue et al. 1993; Teepe et al. 1993a; Brychta et al. 1995, Rivas-Torres et al. 1996; Carsin et al. 2000; Hiroko et al. 2001; Koller et al. 2002; Chester et al. 2004), but also in dermatology (Roseeuw et al. 1989; Teepe et al. 1990; De Luca et al. 1992; Marcusson et al. 1992; Teepe et al. 1993b; Lindgren et al. 1998; Beele et al. 2005), otology (Somers et al. 1997a; Somers et al. 1997b) and stomatology (Tsai et al. 1997). Unfortunately, due to wide patient-to-patient variation and the lack of controlled clinical research, it is currently difficult to tell whether there is a place for cultured epithelial grafts in the treatment of major burns (Wood et al. 2006). Nevertheless, since 1987, cultured epithelial grafts were produced by the Keratinocyte Bank of the Queen Astrid Military Hospital and used, with some success, to accelerate the healing of burns and chronic wounds in more than 1,000 patients (Kets et al. 1991; Duinslaeger et al. 1996, 1997a, b). Taking into account the scarcity of robust scientific evidence, our personal clinical experience and concerns relating to cost-effectiveness, we decided to rationalize the use of keratinocyte grafts in our hospital. Cultured epithelial allografts are now mostly used in the burn centre to stimulate the healing of donor sites and in the chronic wound care centre to treat hard-to-heal venous leg ulcers. It was shown that in our centre, keratinocyte allografts halved the healing time, compared to Op-site treatment, of donor sites (Duinslaeger et al. 1997b), which allowed more efficient autografting (higher grafting frequency and/or smaller skin autograft mesh sizes). Additionally, the donor site surface is most often significantly smaller than the burned surface and thus requires less keratinocyte allografts.
To provide patients with cultured epithelial grafts, a hospital usually needs to approach a specialist tissue establishment, which is accredited by the National Competent Authorities. Since the introduction in March 2004 of the European Union Tissues and Cells Directive (EUTCD—Directive 2004/23/EC) tissue banking regulation has become more stringent, not only with respect to the quality and safety of the source material, but also with respect to the processing environment and the quality and suitability of the products and reagents used in the production process. As a result activities such as testing, validation, traceability, specification and verification processes and risk analysis become integral to operations. To manage and follow up all these different requirements in a structured and unambiguous way, implementing a Quality Management System (QMS) has become best operational practice and is often mandatory by national law (e.g. Belgian transposition of the EUTCD).
The use of Neonatal Foreskin Keratinocytes (NFKs) in a defined and animal product-free cell culture environment has allowed for the development of an efficient allograft production system with an excellent safety profile. Epidermal keratinocytes isolated from neonatal foreskin tissue exhibited tremendous growth potential, allowing predictable subculturing schemes, resulting in increased consistency of final allograft. The establishment of an allogeneic cell banking system enabled extensive safety testing and characterisation of the NFKs in addition to the mandatory tests (for both autologous and allogeneic donors) performed on a blood sample taken from the donor’s biological mother.
All initial cell banking procedures were compliant with Good Manufacturing Practices (GMP) guidelines. The current allograft production is compliant with the recent (December 2009) Belgian transposition of the EUTCD and ISO 9001 QMS requirements. Non-exhaustive examples of measures that have improved safety and quality in keratinocyte processing include: the use of products that meet United States Pharmacopeia (USP) or European Pharmacopeia (EP) specifications, or have a CE mark; the introduction of controlled air environments, and routine in-process controls and Quality Control (QC) testing.
Batch productions (typically 60 units) of cryopreserved allografts, which can be made available off-the-shelf, avoid the long waiting times (3–4 weeks) and difficulties of timing typically experienced with cultured epithelial autografts. In addition, we found cryopreserved allografts to be as effective as cultured epithelial autografts in in vivo wound healing. In our hands, the adhesion or ‘take’ of the cultured epithelial autografts on full thickness burn wounds was disappointing and the lack of a mature dermo-epidermal junction often resulted in mechanically instable grafts. Cultured epithelal allografts were shown to significantly accelerate the healing of donor sites and meshed skin autograft covered burn wounds (Duinslaeger et al. 1997a, b). Finally, the batch production of cryopreserved NFK derived allografts offer manufacturing cost-efficiencies compared with individual, small-scale autologous culturing of adult keratinocytes.
This paper describes in detail our current (i.e. December 2010) cryopreserved allograft production processes.
Materials and methods
Safety of raw materials, media and reagents
To guarantee that all used materials were conforming to applicable EUTCD safety and quality requirements, specification sheets were prepared and controlled through the QMS. Upon receipt of the materials, purchase verification was performed using the available specification sheets. When applicable, EDQM (European Directorate for the Quality of Medicines and Healthcare) certificates of suitability, certificates of analysis, sterility and of origin and health were requested and filed. For materials and reagents that come into contact with the cells during culturing, production and packaging, the highest quality level was used: USP or EP grade for solid substances, GMP grade (or its equivalent, e.g. for “Further Manufacturing Use”) for reagents and solutions, and CE marking for packaging materials. All the media and supplements that were used in the culturing steps were of defined composition and known origin, usually plant, microbial or synthetic. Verification of the suitability of the materials was achieved using stringent quality and safety acceptance criteria.
Donor screening
The following exclusion criteria were applied prior to acceptance and handling of the neonatal foreskin samples:
-
The donor infant’s mother is known to be infected with Human Immunodeficiency Virus (HIV), Hepatitis B Virus (HBV), Hepatitis C Virus (HCV), Human T-Lymphotropic Virus (HTLV), Cytomegalovirus (CMV), syphilis, Epstein-Barr Virus (EBV), or is suspected of having Creutzfeldt-Jakob disease;
-
The mother has a malignancy, a history of malignancy, a systemic disease, which requires chronic medical treatment, or a history of alcohol or drug abuse;
-
The mother received any non-registered drug or drug under investigation during the 4 weeks preceding delivery;
-
The circumcision site shows clinical signs of infection;
-
The donor infant has a congenital malformation, a malignancy, or skin disorder.
Peripheral blood samples were obtained, at the date of birth and according to Good Clinical Practice (GCP), from the donor’s biological mother. Serum was separated from the blood, aliquoted, and used for serological testing (Table 1). A new blood sample was taken at least 6 months later to retest (seroconversion) for HIV, HBV and HCV.
Neonatal foreskin samples
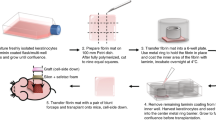
Neonatal foreskin samples were obtained after routine circumcision of newborn males. Informed consent was obtained from the child’s mother. The foreskin tissue was removed according to GCP and conventional aseptic techniques. Collected samples were transferred to 50 ml of basal Epilife® medium (Cascade Biologics, now Gibco, Invitrogen Corporation, USA—a fully defined culture medium with no serum or non-defined tissue extracts), to which 200 IU/ml penicillin, 200 μg/ml streptomycin and 0.5 μg/ml amphotericin B (antibiotic/antimycotic 100×, Gibco, Invitrogen Corporation) were added, and transported at 2–8°C to the processing facility.
Primary isolation of epithelial cells
Upon receipt, the exterior of the specimen container was wiped with 70% isopropanol and placed in a E.C. class A (class 100 according to U.S. Fed. Standard 209) Biological Safety Cabinet (BSC). The circular foreskin sample was transferred to a sterile 90 mm tissue culture dish and cut across its width and flattened. The amount of tissue was estimated in cm². 15 ml Phosphate Buffered Saline (PBS, Lonza) was added to the dish and the tissue was agitated with forceps to rinse it. The rinsing process was then repeated. The skin specimen was then transferred, epidermal side up, to the sterile lid of a 90 mm dish (BD Biosciences). Using curved scissors, subcutaneous fat and reticular dermis were dissected away, leaving an intact papillary dermis. The skin was then washed again in a new dish containing 15 ml PBS and placed on a clean sterile lid of a 90 mm dish and the tissue was cut into strips of approximately 0.5 cm × 1 cm (3–5 strips per foreskin sample, Fig. 1). The tissue strips were transferred to the bottom of a 50 ml conical centrifuge tube (BD Biosciences), which was labelled with the donor code and contained 10 ml of a 25 U/ml dispase II solution (Bacillus polymyxa, Roche Diagnostics) with 200 IU/ml penicillin, 200 μg/ml streptomycin and 0.5 μg/ml amphotericin B. Ensuring that each piece of tissue was submerged in the dispase solution, the tube was securely capped and incubated in a refrigerator for 16–18 h at 2–8°C.

A neonatal foreskin (source of NFK-5) cut into 5 strips each approximately 0.5 cm × 1 cm
The 50 ml conical tube containing the skin pieces in dispase solution was retrieved from the refrigerator and placed in a BSC. Skin pieces and dispase solution were gently poured into a 90 mm petridish. The epidermis was separated from the dermis by holding the dermal side of the skin with forceps and the edge of the epidermis with a second forceps and then pulling both skin components apart. All obtained epidermal pieces were transferred to a sterile 50 ml tube to which 10 ml of pre-warmed (37°C) Trypsin-Versene (porcine, Lonza) was added and incubated in a water bath at 37°C for 30 min, after which the epidermal pieces were torn into smaller fragments using forceps. The solution became cloudy as the individual epidermal cells were released. Immediately thereafter, 20 ml of 0.025% trypsin inhibitor (soybean, Sigma–Aldrich) was added and the solution was pipetted up and down vigorously (approximately 10 times) to further release the epidermal cells. The cell suspension was passed through a 70 μm cell strainer (BD Biosciences), which was positioned on top of a 50 ml conical tube, to remove the remaining cell clusters and pieces of epidermis. The strainer was subsequently washed with 2 ml trypsin inhibitor. The 50 ml conical tube with the epidermal cell suspension was centrifuged for 10 min at 180 g. After centrifugation, the cells were resuspended in 10 ml complete EpiLife® medium, i.e. basal EpiLife® medium supplemented with defined growth supplement S7 (Gibco, Invitrogen Corporation), and homogenised by pipetting up and down. A 20 μl-aliquot of the cell suspension was taken from the tube and diluted 1:1 with a 0.5% trypan blue solution (Biochrom) in an Eppendorf tube. The diluted sample was pipetted in a Bürker haemocytometer and the total number of viable cells was counted. The total amount of primary isolated cells was then cryopreserved in Synth-a-Freeze® cryopreservation medium (Cascade Biologics, now Gibco, Invitrogen Corporation, USA), a DMSO-based cryopreservation solution, at a final concentration of approximately 5 × 105 cells/ml in 2 ml cryovials (Nunc). These vials comprise the Primary Cell Bank (PCB).
Epithelial cell culture
The primary isolated epidermal cells were placed in culture, expanded and banked into a Cell Storage Banking System (CSBS) comprising a Master Cell Bank (MCB), a Master Working Cell Bank (MWCB) and a Working Cell Bank (WCB) (Fig. 2). A cell bank is a collection of vials of uniform composition stored under defined conditions and each containing an aliquot of a single pool of cells. The MCB is the cell bank that is closest to the initially isolated cells. The WCB is derived by expansion of one or more vials of the MCB. The cell culturing history and establishment of the CSBS is described in detail below. The growth potential of the NFKs was evaluated under different conditions to determine defined subculturing schemes. All banking procedures were validated and performed according to standard operating procedures and GMP guidelines.

Diagram of the cell storage bank system (CSBS)
Initiation of primary cultures in a defined animal product-free medium
Primary isolated epidermal cells were seeded in tissue culture flasks (T-flasks, Falcon, BD Biosciences) at a density of 10,000–20,000 cells per cm². The tissue culture flasks were additionally coated with a collagen type-1 solution (PureCol™, Advanced Biomatrix, diluted to 1 μg/ml in PBS), for at least 2 h—but no longer than 24 h, at 37°C. The cell culture medium was complete EpiLife® medium, as described previously. After a maximum of 20 days of primary culture and visual assessment using phase contrast microscopy, cultures consisting of cloning cells that reached at least 30% confluence 20 days after seeding, were passaged to a secondary culture. Cultures exhibiting less than 30% confluence were discarded. Figure 3 shows typical microscopic views of NFKs at different stages of this initiation phase.

Typical microscopic view (magnification 40×) of neonatal foreskin keratinocytes at different stages of the initiation phase. a Initiation of NFK cells, day 8 after seeding. b Growing colonies at day 11. c Colonies at day 15. d Confluent area at day 20
Serial propagation in defined animal product-free medium
When cultured cells reached an acceptable confluence (80–95%), they were passaged from the primary culture to the subsequent culture (serial propagation) until the cells reached the end of their culture lifespan, or until it became obvious that there was no need to continue serial subculturing. Cell density and cumulative population doublings were calculated at the end of each propagation. Cultures that were ready for subculturing were washed once with PBS and then trypsinised with Trypsine-Versene (porcine, Lonza) for 5–10 min at 37°C. The detachment of the cells was observed under a phase contrast microscope and trypsin was inactivated with an equal amount of defined trypsin inhibitor (soybean, Sigma–Aldrich). Cells were homogenised by pipetting up and down, collected in a 50 ml tube and centrifuged for 10 min at 220 g. The pelleted cells were resuspended in complete EpiLife® medium (10 ml per trypsinised tissue culture flask). Homogenisation was again performed by pipetting up and down with the pipette tip at the bottom of the tube. A 100 μl-aliquot of the cell suspension was taken from the tube and diluted 1:1 with a 0.5% trypan blue solution in an Eppendorf tube. The diluted sample was pipetted in a Bürker haemocytometer and the total number of viable cells was counted. The cells were then reseeded in collagen coated tissue culture flasks with complete EpiLife® medium, at a density of 5,000 cells per cm². The flasks were incubated at 37°C in 5% CO2 and a relative humidity greater than 95%.
The confluency of the cell cultures was checked on a regular basis under the microscope and the medium was refreshed every 2–3 days to confluency. At confluence, the cells were subcultured and at the end of passage 2, 4 and 6 the cells were cryopreserved to establish the MCB, MWCB and WCB, respectively.
Establishment of an MCB
To establish the MCB, approximately 5 × 105 viable primary keratinocytes (one vial) were amplified by three culture passages, which represent approximately 9 population doublings (one passage is defined as one cell seeding, the primary culture is considered passage ‘0’). Confluent passage 2 cells were trypsinised, pooled and counted as described above (‘Serial propagation in defined animal product-free medium’). The pooled suspension was centrifuged a second time and the pellet was homogenised in Synth-a-Freeze® cryopreservation solution and transferred to cryovials (2 × 106 cells in 1 ml per vial). The vials were frozen (−1°C/min to −50°C followed by −50°C/min to −150°C) using a controlled rate freezer (Planer, Kryo10, series II) and stored in the vapour phase of liquid nitrogen (<−135°C) in a dedicated vessel equipped with automatic level and temperature control. Since absolute sterility could not be guaranteed for the initial foreskin samples, all culturing steps related to the manufacturing of the MCB were performed in culturing medium supplemented with antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B). Before freezing, the antibiotics were removed by centrifugation and resuspension of the cell pellet in antibiotic-free Synth-a-Freeze® cryopreservation medium.
Establishment of an MWCB
For the establishment of an MWCB, five MCB vials were thawed and propagated for two passages, which corresponds to approximately seven additional population doublings. The seeding density in each passage was 5,000 cells/cm². Confluent passage four cultures were trypsinised, pooled and counted as described above (‘Serial propagation in defined animal product-free medium’). The pooled suspension was centrifuged a second time and the pellet was homogenised in Synth-a-Freeze® cryopreservation solution and transferred to cryovials (5 × 106 cells in 1 ml per vial). The vials were frozen and stored as described above (‘Establishment of an MCB’). No antibiotics were used in establishing the MWCB.
Establishment of a WCB
The WCB contains the source material for the allograft production and was derived from the MWCB. For the establishment of this WCB, 1 MWCB vial was thawed and propagated through two passages (approximately 7 additional population doublings). The resulting cells were transferred to cryo-vials, frozen and stored as described above (‘Establishment of an MCB’). No antibiotics were used in the preparation of the WCB.
Production of Allografts
The production of allografts is performed under aseptic conditions in a class A BSC. The allograft production protocol is described in detail below. One WCB vial is thawed and seeded at approximately 5,000 cells/cm² in collagen-coated (PureCol™, Advanced Biomatrix) 175 cm² tissue culture flasks (BD Biosciences). Cells are propagated until confluence in complete EpiLife® medium in 5% CO2 atmosphere and 95% humidity incubator at 37°C. The propagation takes 4 days and results in approximately 3.5 population doublings. Cells are then detached from the culturing flasks with Trypsine-Versene (porcine, Lonza) and subcultured into new collagen-coated 175 cm2 tissue culture flasks. Cells are cultured until just reaching confluency in complete EpiLife® medium at 37°C. This typically takes an additional 4 days and results in approximately 3.5 population doublings. At confluence, the culture medium is removed and the cultured cells are prepared for harvesting, packaging and freezing.
The final form of the keratinocyte allografts can be viable subconfluent sheets (Fig. 4a) or viable cell suspensions for spray applications (Fig. 4b).

a A plasma coated silicone membrane supports subconfluent keratinocyte growth. Perforations in the substrate allow drainage of wound exudates. b A mix of a viable keratinocyte suspension and fibrin glue are sprayed onto an uneven burn wound bed
The cells can be removed from the culture flasks, using Trypsin-Versene (as described before) and seeded at a density of 13 × 106 cells/cm² on silicone membrane coated with plasma polymerised acylic acid and collagen (PureCol™, Advanced Biomatrix) in a 10 × 10 cm petri dish (Sarstedt), cultured for 24 h at 37°C, and transferred to a custom blister pack (Charpak Ltd) to which defined cryopreservation medium is added (Synth-a-Freeze®, 15 ml). This blister pack is then packaged in a custom laminated aluminium foil pack (Oliver Medical) and cryopreserved using a controlled rate (<1°C/min to −80°C) freezing process. After freezing, the allograft packs are transferred to −80°C freezers for medium term (6 months) storage. The silicone membrane cell delivery technology has been developed by the University of Sheffield and Altrika Ltd in the UK. Full preparation details are outside the scope of the current review, and are described in details elsewhere (Haddow et al. 2006).
Cell suspensions that will be used for spray applications are transferred to cryovials, frozen and stored as described above (‘Establishment of a WCB’). For application on the wound bed, cell suspensions were thawed and washed in PBS and were co-sprayed with fibrin sealant (Tissucol® Kit, Baxter AG) at approximately 50,000 cells/cm2, using the Tissomat® device (Baxter AG). Cell proliferation rate was not significantly reduced when spraying at maximum 20 psi.
Results
Selection of potent and safe donor cells
Epidermal cells derived from the neonatal foreskins of donors that met the safety criteria were first tested for growth potential, to select donor cells with a culture lifespan that is acceptable for long term reproducible processing. The main evaluation criterion was the cumulative population doubling; only the cells with a population doubling of at least 25 were further evaluated. Keratinocytes were isolated from 10 different human neonatal foreskins and subcultivated to assess their total culturing potential. Table 2 shows the results of the growth potential analysis of these 10 human neonatal foreskins biopsies. Initiation of cell growth was not successful for the NFK-7 isolate and isolate NFK-10 was not tested beyond the 4th subculture level because of the relatively long culture time to reach confluence and the low multiplication factor in each subculture, which were an indication of inferior quality and growth potential. The remaining 8 isolates all reached at least 9 subculture levels, which in theory would give rise to more than 1 km² of cultured monolayer.
Cells exhibiting a satisfactory growth potential were cultured under controlled environmental conditions, which were in line with GMP requirements, to create cell banks at different levels (MCB, MWCB and WCB). The banked cells themselves were then tested according to international standards (The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; ICH Topic Q5A “Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin”). The tests that were performed and their corresponding results are shown in Tables 3 and 4. Isolate NFK-1 exhibited the highest cumulative population doubling, but was finally excluded due to positive CMV serology. Isolate NFK-5 was selected for long-term production of keratinocyte allografts.
Establishment of a (quality) controlled culturing scheme
Growth rates and maximal life span of the selected NFK-5 cells were analysed in detail (Table 5) and, as expected, the life span was limited. This conclusion was based on a decreasing proliferation rate. As shown in Table 5, the growth rate remains very consistent until passage 8: seeding 5,000 cells/cm2 systematically resulted into a tenfold increase in cells after 4 days (equivalent to approximately 3.5 population doublings). From passage 9 onwards, the culturing time between subsequent passages increased and the multiplication factor decreased significantly. In addition, the relative proportion of large, non-proliferative cells also increased significantly after passage 8. Light microscopy revealed an increase in senescent and enlarged cells. Furthermore, higher passage (>8) confluent cell cultures harboured less cells than lower passage cultures, and exhibited a higher ratio of viable cells <30 μm and non-viable cells >30 μm (Table 6).
Based on the growth potential evaluation analysis of culturing time and visual cell morphology, passage 8 was selected as the end point for the allografts.
A MCB, MWCB and WCB of NFK-5 cells in respectively, passages 2, 4 and 6 were set up. Once the cell banks were established, the initial growth tests were repeated using the banked NFK-5 cells. The initial growth curve was reproduced with the banked cells (Fig. 5).

Comparison between the growth curves of NFK-5 cells in continuous culture and in a banking system
All manipulations, from donor skin harvesting to final allograft delivery, were described in our quality system procedures and performed by trained operators. These procedures included detailed instructions, actions, and guidance relating to deviations from expected in-process control parameters. These parameters were defined by passage number, seeding density, time to confluence, cell viability and cell morphology. Deviations were evaluated historically, investigated and the possible impact on the safety or quality of the end product assessed.
Consistent allograft productions
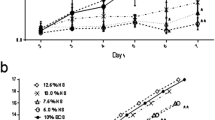
Many allograft productions, containing cells at passage 8, have been initiated from the NFK-5 WCB. Analysis of the growth characteristics of the NFK-5 cells resulted in the cell behaviour under specific conditions being well known. This meant that reproducible production schemes could be established, with a definable end point and specific quality criteria for the final allografts. The culturing conditions were consistent for eight consecutive production runs, resulting in reproducible end products. Figure 6 shows the consistency of these eight consecutive production runs with banked NFK-5 cells and their relation to the initial test curve.

Overview of 8 different NFK-5 production runs and comparison with the initial growth test curve
Animal product-free environment
The goal for optimally safe and consistent cell culture is to use fully defined and animal product-free products. Over the years, we systematically replaced animal products with non-animal alternatives. In this context it is important to note that in the course of 2009 porcine trypsin (Trypsine-Versene, Lonza), which was frequently used throughout the production process, was replaced with recombinant ‘trypsin like dislodging enzyme’ (TrypLE Select, Gibco, Invitrogen Corporation). Today, some of our epithelial allograft delivery formulations using NFKs still require the use of animal-derived collagen as a culture substrate.
Discussion
In 1987, the Keratinocyte Bank of the Queen Astrid Military Hospital started producing cultured epithelial autografts using the method of Rheinwald and Green. This method uses keratinocytes derived from adult skin, feeder layers of mitomycin C growth arrested mouse fibroblasts and serum-containing media. Since then the culturing system has been updated to accommodate scientific and regulatory advances. Figure 7 shows a detailed history of the changes introduced to our epithelial graft production system.

History of all the changes that were introduced in the cultured epithelial allograft production system
The first major change was implemented in 1989, when we gradually switched from autologous (Phase 1) to allogeneic source material (Phase 2), which resulted in efficient production planning and consequently an improved availability of epithelial grafts for surgery.
A second advance enabled the production of cryopreserved allografts (Phase 3), which allowed off-the-shelf use and pre-transplantation microbial testing.
The next major change was the removal of the mouse fibroblast feeder layer from the culturing system and, shortly thereafter the introduction of NFKs as source material (Phase 4). Mouse fibroblast feeder layers support the growth of human keratinocytes, but there is a risk that they could introduce remnants of animal components (e.g. mouse DNA) into the epithelial allografts. The grafts would then be defined as xenografts from a regulatory viewpoint. The introduction of NFKs, in turn, increased the consistency and the performance of our culturing system. In contrast to the irregular, unpredictable growth rates and variable end points associated with the culture of adult human epidermal keratinocytes (HEKas), NFKs showed regular and consistent growth for up to 50 population doublings, even after more than 5 years of cryopreservation, which is comparable to established cell lines. This tremendous proliferation potential allowed the use of more defined subculturing schemes. HEKas were also initiated and propagated in our culturing system, but they never reached the same culture level as the NFKs. Coolen et al. (2007) reported similar results for HEKas in a feeder layer free culturing system. In addition, the establishment of an NFK cell bank system increased safety, because it enabled extensive safety testing of the cells themselves in addition to the usual tests that are traditionally performed on the blood of the donor. In addition, as the cell source remains unchanged for several years and numerous allograft batches, their safety profile for patients increases because the clinical use of these allografts is documented for hundreds of applications. The use of the same cell source for many years also facilitates the traceability of the allografts. Finally, the use of banked cell stocks for multiple allograft productions reduced the risk of mutations associated with serial cultivation.
Braye et al. (2000) developed a cell bank system for adult allogeneic keratinocytes, but this approach has only limited possibilities, mainly due to the constant need for new donor cells, which results in repeated safety testing and variations in growth potential of the starting cells. Our NFK cell banks were tested for several viral agents, mycoplasmas, tumorigenicity and karyology. The financial challenges associated with performing these tests for keratinocytes derived from multiple adult donors is considerable.
In our system, the use of HEKas would lead to substantially higher allograft production costs, due to higher operational and safety testing costs. Moreover, it may not be possible to reach NFK safety and quality levels with banked HEKas, which usually originate from donors that are relatively hard to screen for safety (e.g. deceased donors and surgical waste donors). Finally, HEKa allograft consistency would also be compromised because donor age and anatomical site of skin harvesting can influence the quality and the life span of the cultured cells (data not shown). NFK allograft consistency is likely to have a positive influence on the graft success rate.
Phase 5 saw the introduction of a totally defined culturing medium and an ISO 9001 certified QMS, which led to a further improvement of allograft quality, safety and consistency. The use of controlled environment and products that meet USP and/or EP specifications or have a CE mark, in process controls and QC testing are a few examples of the measures that improved the safety and quality of our allografts. In addition, all media and reagents that came in contact with the cells and the end product were produced in compliance with GMP by specialist manufacturers.
In Phase 6, which at the time of writing is ongoing, we aim to progress to a fully defined and animal product-free cell culture environment, which avoids the possible transmittance of known and unknown animal infectious agents (e.g. scrapie and bovine spongiform encephalopathy) to workers exposed to the cell culture media and reagents and patients treated with allografts (Sun et al. 2004). At present the NFK delivery formulations, which are clinically available and minimise the use of animal derived components, are cell suspensions and subconfluent cells seeded on a silicone membrane. The latter presentation still requires the use of animal-derived collagen but evaluation of a human recombinant Type I collagen (Coating Matrix Kit, Gibco, Invitrogen Corporation) forms the focus of current work in this laboratory. Whilst subconfluent cells are acknowledged to provide optimum mitogenic activity, the mode of action for allogeneic cells compared to autologous cells is quite different. Autologous grafts will provide permanent cover for burns patients, and they were developed as complete cell sheets for this purpose. One of the features of keratinocytes is that they display a tightly regulated proliferation/differentiation behaviour. One of the triggers for differentiation is cell-to-cell contact. Integrated cell sheets are therefore differentiated cells which can provide a barrier function but are not as potent mitogenically as subconfluent cells. The silicone membrane delivery technology described in this publication and elsewhere was developed by the University of Sheffield and Altrika as a way of delivering subconfluent cells whilst also providing a wound cover. The approach was initially developed for use with autologous cells, but has since been adapted to deliver allogeneic NFKs. One of the potential drawbacks with delivering subconfluent allogeneic cells is related to their mode of action. In contrast to autologous cells, which are capable of permanent take on a wound, allogeneic cells will be rejected over time, albeit with minimal inflammatory response and no requirement for immunosuppressant drugs. Their benefit to burns patients is in providing accelerated healing by potent mitogenic stimulation of remnant cells in (burn) wounds or donor sites. In such cases it may be desirable to increase the numbers of allogeneic cells being delivered to a particular wound site, in effect increasing the mitogenic dose. If the numbers of subconfluent cells are increased, eventually they will reach confluency and the defined differentiation pathway will be activated. There is therefore a balance between cells displaying optimum mitogenic activity in a subconfluent state, and increasing mitogenic activity by having higher cell numbers, but in a differentiated state. The clinical outcomes of the two approaches have, unfortunately, not yet been evaluated in a controlled manner. To generate an intact sheet of NFKs, and build up cell numbers, stratification of cells is required and previously this has required the use of a serum-containing media. Allografts produced in an animal-free stratification system, containing animal product-free soy peptone (BBL™ Phytone™ Peptone, BD Biosciences), are currently being validated. The relatively high cost and limitations on therapeutic use of some of the animal product-free alternatives could delay routine clinical application of these Phase 6 allografts. Some manufacturers do not wish to disclose the exact composition of proprietary media or adjuvants and the clinical indemnity issues of using these products in patients are such that some of these promising media are currently not licensed for such use (Bullock et al. 2006).
In summary, quality controlled batch productions of consistent allografts have been cryopreserved for off-the-shelf use. The developments outlined above have significantly increased the performance, usability, quality and safety of our allografts. Depending on the wound type, treatment regime, surgical techniques and clinical timing, specific NFK allograft delivery formats can be prepared. As such, we are now focussing on the development and comparison of additional allograft delivery formats, such as animal-free multilayer sheets, monolayer cultures on silicone substrates, lyophilised keratinocytes and composite living skin equivalents.
References
Beele H, de la Brassine M, Lambert J, Suys E, De Cuyper C, Decroix J, Boyden B, Tobback L, Hulstaert F, De Schepper S, Brissinck J, Delaey B, Draye JP, De Deene A, De Waele P, Verbeken G (2005) A prospective multicenter study of the efficacy and tolerability of cryopreserved allogenic human keratinocytes to treat venous leg ulcers. Int J Low Extrem Wounds 4:225–233
Bolivar-Flores J, Poumian E, Marsch-Moreno M, Montes de Oca G, Kuri-Harcuch W (1990) Use of cultured human epidermal keratinocytes for allografting burns and conditions for temporary banking of the cultured allografts. Burns 16:3–8
Braye F, Pascal P, Bertin-Maghit M, Colpart JJ, Tissot E, Damour O (2000) Advantages of using a bank of allogeneic keratinocytes for the rapid coverage of extensive and deep second-degree burns. Med Biol Eng Comput 38:248–252
Brychta P, Sucánek I, Rihova H, Adler J, Komárková J (1995) Cultured epidermal allografts for the treatment of deep dermal burns. Acta Chir Plast 37:20–24
Bullock AJ, Higham MC, MacNeil S (2006) Use of human fibroblasts in the development of a xenobiotic-free culture and delivery system for human keratinocytes. Tissue Eng 12:245–255
Burt AM, Pallet CD, Sloane JP, O’Hare MJ, Schafler KF, Yardeni P, Eldad A, Clarke JA, Gusterson BA (1989) Survival of cultured allografts in patients with burns assessed with probe specific for Y chromosome. Br Med J 298:915–917
Carsin H, Ainaud P, Le Bever H, Rives JM, Lakhel A, Stephanazzi J, Lambert F, Perrot J (2000) Cultured epithelial autografts in extensive burn coverage of severely traumatized patients: a five year single-center experience with 30 patients. Burns 26:379–387
Chester DL, Balderson DS, Papini RP (2004) A review of keratinocyte delivery to the wound bed. J Burn Care Rehabil 25:266–275
Compton CC, Regauer S, Seiler GR, Landry DB (1990) Human Merkel cell regeneration in skin derived from cultured keratinocyte grafts. Lab Invest 63:233–241
Coolen NA, Verkerk M, Reijnen L, Vlig M, van den Bogaerdt AJ, Breetveld M, Gibbs S, Middelkoop E, Ulrich MM (2007) Culture of keratinocytes for transplantation without the need of feeder layer cells. Cell Transplant 16:649–661
De Luca M, Albanese E, Bondanza S, Megna M, Ugozzoli L, Molina F, Cancedda R, Santi PL, Bormioli M, Stella M, Magliacani G (1989) Multicenter experience in the treatment of burns with autologous and allogenic cultured epithelium, fresh or preserved in a frozen state. Burns 15:303–309
De Luca M, Albanese E, Cancedda R, Viacava A, Faggioni A, Zambruno G, Giannetti A (1992) Treatment of leg ulcers with cryopreserved allogeneic cultured epithelium. Arch Dermatol 128:633–638
Directive 2004/23/EC of the European Parliamant and of the Council of 31 March 2004 on setting standards of quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human tissues and cells
Duinslaeger L, Verbeken G, Reper P, Delaey B, Vanhalle S, Vanderkelen A (1996) Lyophilized keratinocyte cell lysates contain multiple mitogenic activities and stimulate closure of meshed skin autograft-covered burn wounds with efficiency similar to that of fresh allogeneic keratinocyte cultures. Plast Reconstr Surg 98:110–117
Duinslaeger L, Delaey B, Vanderkelen A (1997a) Short-and long-term results of application of allogeneic cultured keratinocytes on burn wounds and burn scar. Eur J Plast Surg 318:1–5
Duinslaeger L, Verbeken G, Vanhalle S, Vanderkelen A (1997b) Cultured allogeneic keratinocyte sheets accelerate healing compared to Op-Site treatment of donor sites in burns. J Burn Care Rehabil 18:545–551
Fratianne R, Papay F, Housini I, Lang C, Schafer IA (1993) Keratinocyte allografts accelerate healing of split-thickness donor sites: applications for improved treatment of burns. J Burn Care Rehabil 14:148–154
Haddow D, MacNeil S, Short R (2006) A cell therapy for chronic wounds based upon a plasma polymer delivery surface. Plasma Processes Polym 3:419–430
Herzog SR, Meyer A, Woodley D, Peterson HD (1988) Wound coverage with cultured autologous keratinocytes: use after burn wound excision, including biopsy followup. J Trauma 28:195–198
Hiroko Y, Yukihiro U, Toshihiko Y, Misa Y, Kensuke K, Yojiro I, Yoshiaki T (2001) Cryopreserved cultured epidermal allografts achieved early closure of wounds and reduced scar formation in deep partial-thickness burn wounds (DDB) and split-thickness skin donor sites of pediatric patients. Burns 27:689–698
Kets E, Paye M, Verbeken G, Coopman P, Calders P, Dimitrieff A, Vanderkelen A, Roseeuw D (1991) Treatment of burn wounds with cultured epidermis. Ann Med Milit Belg 5:5–10
Koller R, Bierochs B, Meissl G, Rab M, Frey M (2002) The use of allogeneic keratinocytes for the early coverage of deep dermal burns–indications, results and problems. Cell Tissue Bank 3:11–14
Lindgren C, Marcusson JA, Toftgard R (1998) Treatment of venous leg ulcers with cryopreserved cultured allogeneic keratinocytes: a prospective open controlled study. Br J Dermatol 139:271–275
Madden MR, Finkelstein JL, Staiano-Coico L, Goodwin CW, Shires GT, Nolan EE, Hefton JM (1986) Grafting of cultured allogeneic epidermis on second- and third-degree burn wounds on 26 patients. J Trauma 26:955–962
Marcusson JA, Lindgren C, Berghard A, Toftgard R (1992) Allogeneic cultured keratinocytes in the treatment of leg ulcers. Acta Derm Venereol 72:61–64
O’Connor NE, Mulliken JB, Banks-Schlegel S, Kehinde O, Green H (1981) Grafting of burns with cultured epithelium prepared from autologous epidermal cells. Lancet 317:75–78
Phillips TJ, Provan A, Colbert D, Easly KW (1993) A randomized single-blind controlled study of cultured epidermal allografts in the treatment of split-thickness skin graft donor sites. Arch Dermatol 129:879–882
Rheinwald JG, Green H (1975) Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6:331–343
Rheinwald JG, Green H (1977) Epidermal growth factor and the multiplication of human epidermal keratinocytes. Nature 265:421–424
Rivas-Torres MT, Amato D, Arámbula-Alvarez H, Kuri-Harcuch W (1996) Controlled clinical study of skin donor sites and deep partial-thickness burns treated with cultured epidermal allografts. Plast Reconstr Surg 98:279–287
Roseeuw D, Lissens W, De Coninck A, Vandenberghe Y, Liesbaers I (1989) Epidermal DNA analysis of chronic ulcers grafted with cultured epidermis. J Invest Dermatol 92:509A
Rue LW 3rd, Cioffi WG, McManus WF, Pruitt BA Jr (1993) Wound closure and outcome in extensively burned patients treated with cultured autologous keratinocytes. J Trauma 34:662–667
Somers T, Verbeken G, Duinslaeger L, Vanhalle S, Govaerts P, Delaey B, Offeciers E (1997a) Treatment of chronic postoperative otorrhea with cultured keratinocyte sheets. Ann Otol Rhinol Laryngol 106:15–21
Somers T, Duinslaeger L, Delaey B, Vanhalle S, Boedts D, Govaerts P, Offeciers E (1997b) Stimulation of epithelial healing in chronic postoperative otorrhea using lyohilized cultured keratinocyte lysates. The Am J Otol 18:702–706
Sun T, Higham M, Layton C, Haycock J, Short R, MacNeil S (2004) Developments in xenobiotic-free culture of human keratinocytes for clinical use. Wound Repair Regen 12:626–634
Teepe RGC, Koebrugge EJ, Ponec M, Vermeer BJ (1990) Fresh versus cryopreserved cultured allografts for the treatment of chronic skin ulcers. Br J Dermatol 122:81–89
Teepe RGC, Koch R, Haeseker B (1993a) Randomized trial comparing cryopreserved cultured epidermal allografts with tulle-gras in the treatment of split-thickness skin grafts donor sites. J Trauma 6:850–854
Teepe RGC, Roseeuw DI, Hermans J, Koebrugge EJ, Altena T, de Coninck A, Ponec M, Vermeer BJ (1993b) Randomized trial comparing cryopreserved cultured epidermal allografts with hydrocolloid dressings in healing chronic venous ulcers. J Am Acad Dermatol 29:982–988
Tsai CY, Ueda M, Hata K, Horie K, Hibino Y, Sugimura Y, Toriyama K, Torii S (1997) Clinical results of cultured epithelial cell grafting in the oral and maxillofacial region. J Craniomaxillofac Surg 25:4–8
Wood FM, Kolybaba ML, Allen P (2006) The use of cultured epithelial autograft in the treatment of major burn injuries: a critical review of the literature. Burns 32:395–401
Acknowledgments
This allograft production protocol is the result of 23 years of research and development to which dozens of researchers and technicians have contributed. Parts of the development were made possible through a close collaboration with several biotechnology companies, but especially with Xcellentis N.V. and Innogenetics N.V. This research was supported by grants from ‘Vlaams Actieprogramma Biotechnologie (VLAB)’, ‘Agentschap voor Innovatie door Wetenschap en Technologie (IWT)’, the ‘Vrije Universiteit Brussel (VUB)’ and Belgian Defence.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
De Corte, P., Verween, G., Verbeken, G. et al. Feeder layer- and animal product-free culture of neonatal foreskin keratinocytes: improved performance, usability, quality and safety. Cell Tissue Bank 13, 175–189 (2012). https://doi.org/10.1007/s10561-011-9247-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10561-011-9247-3